CANCER IS A PREVENTABLE, TREATABLE & REVERSILBLE DIS-EASE!
“The cure for cancer is NOT found in its treatment but is found in its prevention” – Dr. Robert O. Young
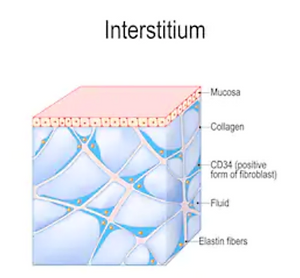
Ninety-five-percent (95%) of ALL sickness and diseases are caused by what you eat, what you drink, what you breath and what you think. Only five-percent (5%) of ALL sickness and diseases are caused by genetics. The five-percent (5%) of All genetic factors are triggered by the epi-genetics or the alkaline environment of interstitial fluids of the Intersitium (the largest organ of the human body) determined by what you eat, what you drink, what you breath, and what you thing, Therefore, one-hundred-percent (100%) is caused by what you eat, what you drink, what you breath and what you think.(Sympathetic Resonance Technology, Scientific Foundations and Summary of Biologic and Clinical Studies, Dec. 2002, Vol. 8, No. 6: 835-842, Alkalizing Nutritional Therapy, medcraveonline.com/IJCAM/IJCAM-02-00046.php Robert O Young and Galina Migalko. Universal Medical Imaging Group, Medical doctor, non-invasive medical diagnostics, USA)

Abstract
This year, more than 1.5 million Americans and more than 18 million people worldwide are expected to be diagnosed with cancer, a disease commonly believed to be preventable. Only 5–10% of all cancer cases can be attributed to genetic defects, whereas the remaining 90–95% have their roots in the environment and lifestyle. The lifestyle factors include cigarette smoking, diet (fried foods, red meat), alcohol, sun exposure, environmental pollutants, infections, stress, obesity, and physical inactivity. The evidence indicates that of all cancer-related deaths, almost 25–30% are due to tobacco, as many as 30–35% are linked to diet, about 15–20% are due to infections, and the remaining percentage are due to other factors like radiation, stress, physical activity, environmental pollutants etc. Therefore, cancer prevention requires smoking cessation, increased ingestion of fruits and vegetables, moderate use of alcohol, caloric restriction, exercise, avoidance of direct exposure to sunlight, minimal meat consumption, use of whole grains, use of vaccinations, and regular check-ups. In this review, we present evidence that inflammation is the link between the agents/factors that cause cancer and the agents that prevent it. In addition, we provide evidence that cancer is a preventable disease that requires major lifestyle changes.

INTRODUCTION
After sequencing his own genome, pioneer genomic researcher Craig Venter remarked at a leadership for the twenty-first century conference, “Human biology is actually far more complicated than we imagine.
Everybody talks about the genes that they received from their mother and father, for this trait or the other. But in reality, those genes have very little impact on life outcomes. Our biology is way too complicated for that and deals with hundreds of thousands of independent factors. Genes are absolutely not our fate. They can give us useful information about the increased risk of a disease, but in most cases they will not determine the actual cause of the disease, or the actual incidence of somebody getting it. Most biology will come from the complex interaction of all the proteins and cells working with environmental factors, not driven directly by the genetic code” (http://indiatoday.digitaltoday.in/index.php?
This statement is very important because looking to the human genome for solutions to most chronic illnesses, including the diagnosis, prevention, and treatment of cancer, is overemphasized in today’s world. Observational studies, however, have indicated that as we migrate from one country to another, our chances of being diagnosed with most chronic illnesses are determined not by the country we come from but by the country we migrate to (1–4). In addition, studies with identical twins have suggested that genes are not the source of most chronic illnesses. For instance, the concordance between identical twins for breast cancer was found to be only 20% (5). Instead of our genes, our lifestyle and environment account for 90–95% of our most chronic illnesses.
Cancer continues to be a worldwide killer, despite the enormous amount of research and rapid developments seen during the past decade. According to recent statistics, cancer accounts for about 23% of the total deaths in the USA and is the second most common cause of death after heart disease (6). Death rates for heart disease, however, have been steeply decreasing in both older and younger populations in the USA from 1975 through 2002. In contrast, no appreciable differences in death rates for cancer have been observed in the United States (6).
By 2020, the world population is expected to have increased to 7.5 billion; of this number, approximately 15 million new cancer cases will be diagnosed, and 12 million cancer patients will die (7). These trends of cancer incidence and death rates again remind us of Dr. John Bailer’s May 1985 judgment of the US national cancer program as a “qualified failure,” a judgment made 14 years after President Nixon’s official declaration of the “War on Cancer.” Even after an additional quarter century of extensive research, researchers are still trying to determine whether cancer is preventable and are asking “If it is preventable, why are we losing the war on cancer?” In this review, we attempt to answer this question by analyzing the potential risk factors of cancer and explore our options for modulating these risk factors.
Cancer is caused by both internal factors (such as inherited mutations, hormones, and immune conditions) and environmental/acquired factors (such as tobacco, diet, radiation, and infectious organisms; Fig. 1). The link between diet and cancer is revealed by the large variation in rates of specific cancers in various countries and by the observed changes in the incidence of cancer in migrating. For example, Asians have been shown to have a 25 times lower incidence of prostate cancer and a ten times lower incidence of breast cancer than do residents of Western countries, and the rates for these cancers increase substantially after Asians migrate to the West (http://www.dietandcancerreportorg/?p=ER).

The role of genes and environment in the development of cancer.
A: The percentage contribution of genetic and environmental factors to cancer. The contribution of genetic factors and environmental factors towards cancer risk is 5–10% and 90–95% respectively.
B: Family risk ratios for selected cancers. The numbers represent familial risk ratios, defined as the risk to a given type of relative of an affected individual divided by the population prevalence. The data shown here is taken from a study conducted in Utah to determine the frequency of cancer in the first-degree relatives (parents + siblings + offspring). The familial risk ratios were assessed as the ratio of the observed number of cancer cases among the first degree relatives divided by the expected number derived from the control relatives, based on the years of birth (cohort) of the case relatives. In essence, this provides an age-adjusted risk ratio to first-degree relatives of cases compared with the general population.
C: Percentage contribution of each environmental factor. The percentages represented here indicate the attributable-fraction of cancer deaths due to the specified environmental risk factor.
The importance of lifestyle factors in the development of cancer was also shown in studies of monozygotic twins (8). Only 5–10% of all cancers are due to an inherited gene defect. Various cancers that have been linked to genetic defects are shown in Fig. 2. Although all cancers are a result of multiple mutations (9, 10), these mutations are due to interaction with the environment (11, 12).

Genes associated with risk of different cancers
These observations indicate that most cancers are not of hereditary origin and that lifestyle factors, such as dietary habits, smoking, alcohol consumption, and infections, have a profound influence on their development (13). Although the hereditary factors cannot be modified, the lifestyle and environmental factors are potentially modifiable. The lesser hereditary influence of cancer and the modifiable nature of the environmental factors point to the preventability of cancer. The important lifestyle factors that affect the incidence and mortality of cancer include tobacco, alcohol, diet, obesity, infectious agents, environmental pollutants, and radiation.
RISK FACTORS OF CANCER
Tobacco
Smoking was identified in 1964 as the primary cause of lung cancer in the US Surgeon General’s Advisory Commission Report (http://profiles.nlm.nih.gov/NN/Views/AlphaChron/date/10006/05/01/2008), and ever since, efforts have been ongoing to reduce tobacco use. Tobacco use increases the risk of developing at least 14 types of cancer (Fig. 3). In addition, it accounts for about 25–30% of all deaths from cancer and 87% of deaths from lung cancer. Compared with nonsmokers, male smokers are 23 times and female smokers 17 times more likely to develop lung cancer.
(http://www.cancer.org/docroot/STT/content/STT_1x_Cancer_Facts_and_Figures_2008.asp accessed on 05/01/2008).
The carcinogenic effects of active smoking are well documented; the U. S. Environmental Protection Agency, for example, in 1993 classified environmental tobacco smoke (from passive smoking) as a known (Group A) human lung carcinogen.
(http://cfpub2.epa.gov/ncea/cfm/recordisplay.cfm?deid=2835 accessed on 05/01/2008).
Tobacco contains at least 50 carcinogens. For example, one tobacco metabolite, benzopyrenediol epoxide, has a direct etiologic association with lung cancer (14). Among all developed countries considered in total, the prevalence of smoking has been slowly declining; however, in the developing countries where 85% of the world’s population resides, the prevalence of smoking is increasing. According to studies of recent trends in tobacco usage, developing countries will consume 71% of the world’s tobacco by 2010, with 80% increased usage projected for East Asia.
(http://www.fao.org/DOCREP/006/Y4956E/Y4956E00.HTM accessed on 01/11/08)
The use of accelerated tobacco-control programs, with an emphasis in areas where usage is increasing, will be the only way to reduce the rates of tobacco-related cancer mortality.
Cancers that have been linked to alcohol and smoking
Percentages represent the cancer mortality attributable to alcohol and smoking in men and women as reported by Irigaray et al. (see 13).
How smoking contributes to cancer is not fully understood. We do know that smoking can alter a large number of cell-signaling pathways. Results from studies in our group have established a link between cigarette smoke and inflammation. Specifically, we showed that tobacco smoke can induce activation of NF-κB, an inflammatory marker (15,16). Thus, anti-inflammatory agents that can suppress NF-κB activation may have potential applications against cigarette smoke.
We also showed that curcumin, derived from the dietary spice turmeric, can block the NF-κB induced by cigarette smoke (15). In addition to curcumin, we discovered that several natural phytochemicals also inhibit the NF-κB induced by various carcinogens (17). Thus, the carcinogenic effects of tobacco appear to be reduced by these dietary agents. A more detailed discussion of dietary agents that can block inflammation and thereby provide chemopreventive effects is presented in the following section.

Alcohol
The first report of the association between alcohol and an increased risk of esophageal cancer was published in 1910 (18). Since then, a number of studies have revealed that chronic alcohol consumption is a risk factor for cancers of the upper aerodigestive tract, including cancers of the oral cavity, pharynx, hypopharynx, larynx, and esophagus (18–21), as well as for cancers of the liver, pancreas, mouth, and breast (Fig. 3). Williams and Horn (22), for example, reported an increased risk of breast cancer due to alcohol. In addition, a collaborative group who studied hormonal factors in breast cancer published their findings from a reanalysis of more than 80% of individual epidemiological studies that had been conducted worldwide on the association between alcohol and breast cancer risk in women. Their analysis showed a 7.1% increase in relative risk of breast cancer for each additional 10 g/day intake of alcohol (23). In another study, Longnecker et al., (24) showed that 4% of all newly diagnosed cases of breast cancer in the USA are due to alcohol use. In addition to it being a risk factor for breast cancer, heavy intake of alcohol (more than 50–70 g/day) is a well-established risk factor for liver (25) and colorectal (26,27) cancers.
There is also evidence of a synergistic effect between heavy alcohol ingestion and hepatitis C virus (HCV) or hepatitis B virus (HBV), which presumably increases the risk of hepatocellular carcinoma (HCC) by more actively promoting cirrhosis. For example, Donato et al. (28) reported that among alcohol drinkers, HCC risk increased linearly with a daily intake of more than 60 g. However, with the concomitant presence of HCV infection, the risk of HCC was two times greater than that observed with alcohol use alone (i.e., a positive synergistic effect). The relationship between alcohol and inflammation has also been well established, especially in terms of alcohol-induced inflammation of the liver.
How alcohol contributes to carcinogenesis is not fully understood but ethanol may play a role. Study findings suggest that ethanol is not a carcinogen but is a cocarcinogen (29). Specifically, when ethanol is metabolized, acetaldehyde and free radicals are generated; free radicals are believed to be predominantly responsible for alcohol-associated carcinogenesis through their binding to DNA and proteins, which destroys folate and results in secondary hyperproliferation. Other mechanisms by which alcohol stimulates carcinogenesis include the induction of cytochrome P-4502E1, which is associated with enhanced production of free radicals and enhanced activation of various procarcinogens present in alcoholic beverages; a change in metabolism and in the distribution of carcinogens, in association with tobacco smoke and diet; alterations in cell-cycle behavior such as cell-cycle duration leading to hyperproliferation; nutritional deficiencies, for example, of methyl, vitamin E, folate, pyridoxal phosphate, zinc, and selenium; and alterations of the immune system. Tissue injury, such as that occurring with cirrhosis of the liver, is a major prerequisite to HCC. In addition, alcohol can activate the NF-κB proinflammatory pathway (30), which can also contribute to tumorigenesis (31). Furthermore, it has been shown that benzopyrene, a cigarette smoke carcinogen, can penetrate the esophagus when combined with ethanol (32). Thus anti-inflammatory agents may be effective for the treatment of alcohol-induced toxicity.
In the upper aerodigestive tract, 25–68% of cancers are attributable to alcohol, and up to 80% of these tumors can be prevented by abstaining from alcohol and smoking (33). Globally, the attributable fraction of cancer deaths due to alcohol drinking is reported to be 3.5% (34). The number of deaths from cancers known to be related to alcohol consumption in the USA could be as low as 6% (as in Utah) or as high as 28% (as in Puerto Rico). These numbers vary from country to country, and in France have approached 20% in males (18).
Diet
In 1981, Doll and Peto (21) estimated that approximately 30–35% of cancer deaths in the USA were linked to diet (Fig. 4). The extent to which diet contributes to cancer deaths varies a great deal, according to the type of cancer (35). For example, diet is linked to cancer deaths in as many as 70% of colorectal cancer cases. How diet contributes to cancer is not fully understood. Most carcinogens that are ingested, such as nitrates, nitrosamines, pesticides, and dioxins, come from food or food additives or from cooking.

Cancer deaths (%) linked to diet as reported by Willett (see 35)
Heavy consumption of red meat is a risk factor for several cancers, especially for those of the gastrointestinal tract, but also for colorectal (36–38), prostate (39), bladder (40), breast (41), gastric (42), pancreatic, and oral (43) cancers. Although a study by Dosil-Diaz et al., (44) showed that meat consumption reduced the risk of lung cancer, such consumption is commonly regarded as a risk for cancer for the following reasons. The heterocyclic amines produced during the cooking of meat are carcinogens.
Charcoal cooking and/or smoke curing of meat produces harmful carbon compounds such as pyrolysates and amino acids, which have a strong cancerous effect. For instance, PhIP (2-amino-1-methyl-6-phenyl-imidazo[4,5-b]pyridine) is the most abundant mutagen by mass in cooked beef and is responsible for ~20% of the total mutagenicity found in fried beef. Daily intake of PhIP among Americans is estimated to be 280–460 ng/day per person (45).
Nitrites and nitrates are used in meat because they bind to myoglobin, inhibiting botulinum exotoxin production; however, they are powerful carcinogens (46). Long-term exposure to food additives such as nitrite preservatives and azo dyes has been associated with the induction of carcinogenesis (47).
Furthermore, bisphenol from plastic food containers can migrate into food and may increase the risk of breast (48) and prostate (49) cancers. Ingestion of arsenic may increase the risk of bladder, kidney, liver, and lung cancers (50). Saturated fatty acids, trans fatty acids, and refined sugars and flour present in most foods have also been associated with various cancers. Several food carcinogens have been shown to activate inflammatory pathways.
Obesity
According to an American Cancer Society study (51), obesity has been associated with increased mortality from cancers of the colon, breast (in postmenopausal women), endometrium, kidneys (renal cell), esophagus (adenocarcinoma), gastric cardia, pancreas, prostate, gallbladder, and liver (Fig. 5).
Findings from this study suggest that of all deaths from cancer in the United States, 14% in men and 20% in women are attributable to excess weight or obesity. Increased modernization and a Westernized diet and lifestyle have been associated with an increased prevalence of overweight people in many developing countries (52).

Various cancers that have been linked to obesity. In the USA overweight and obesity could account for 14% of all deaths from cancer in men and 20% of those in women (see 51).
Studies have shown that the common denominators between obesity and cancer include neurochemicals; hormones such as insulinlike growth factor 1 (IGF-1), insulin, leptin; sex steroids; adiposity; insulin resistance; and inflammation (53).
Involvement of signaling pathways such as the IGF/insulin/Akt signaling pathway, the leptin/JAK/STAT pathway, and other inflammatory cascades have also been linked with both obesity and cancer (53). For instance, hyperglycemia, has been shown to activate NF-κB (54), which could link obesity with cancer. Also known to activate NF-κB are several cytokines produced by adipocytes, such as leptin, tumor necrosis factor (TNF), and interleukin-1 (IL-1) (55). Energy balance and carcinogenesis has been closely linked (53). However, whether inhibitors of these signaling cascades can reduce obesity-related cancer risk remains unanswered. Because of the involvement of multiple signaling pathways, a potential multi-targeting agent will likely be needed to reduce obesity-related cancer risk.
Acidic or So-Called Infectious Agents
Worldwide, an estimated 17.8% of neoplasms are associated with infections; this percentage ranges from less than 10% in high-income countries to 25% in African countries (56, 57). Viruses account for most infection-caused cancers (Fig. 6). Human papillomavirus, Epstein Barr virus, Kaposi’s sarcoma-associated herpes virus, human T-lymphotropic virus 1, HIV, HBV, and HCV are associated with risks for cervical cancer, anogenital cancer, skin cancer, nasopharyngeal cancer, Burkitt’s lymphoma, Hodgkin’s lymphoma, Kaposi’s sarcoma, adult T-cell leukemia, B-cell lymphoma, and liver cancer.

Various cancers that have been linked to infection. The estimated total of infection attributable cancer in the year 2002 is 17.8% of the global cancer burden. The infectious agents associated with each type of cancer is shown in the bracket. HPV Human papilloma virus, HTLV human T-cell leukemia virus, HIV human immunodeficiency virus, EBV Epstein–Barr virus (see 57).
In Western developed countries, human papillomavirus and HBV are the most frequently encountered oncogenic DNA viruses. Human papillomavirus is directly mutagenic by inducing the viral genes E6 and E7 (58), whereas HBV is believed to be indirectly mutagenic by generating reactive oxygen species through chronic inflammation (59–61). Human T-lymphotropic virus is directly mutagenic, whereas HCV (like HBV) is believed to produce oxidative stress in infected cells and thus to act indirectly through chronic inflammation (62, 63). However, other microorganisms, including selected parasites such as Opisthorchis viverrini or Schistosoma haematobium and bacteria such as Helicobacter pylori, may also be involved, acting as cofactors and/or carcinogens (64).
The mechanisms by which infectious agents promote cancer are becoming increasingly evident. Infection-related inflammation is the major risk factor for cancer, and almost all viruses linked to cancer have been shown to activate the inflammatory marker, NF-κB (65). Similarly, components of Helicobacter pylorihave been shown to activate NF-κB (66). Thus, agents that can block chronic inflammation should be effective in treating these conditions.
Environmental Pollution
Environmental pollution has been linked to various cancers (Fig. 7). It includes outdoor air pollution by carbon particles associated with polycyclic aromatic hydrocarbons (PAHs); indoor air pollution by environmental tobacco smoke, formaldehyde, and volatile organic compounds such as benzene and 1,3-butadiene (which may particularly affect children); food pollution by food additives and by carcinogenic contaminants such as nitrates, pesticides, dioxins, and other organochlorines; carcinogenic metals and metalloids; pharmaceutical medicines; and cosmetics (64).

Various cancers that have been linked to environmental carcinogens. The carcinogens linked to each cancer is shown inside bracket. (see 64).
Numerous outdoor air pollutants such as PAHs increase the risk of cancers, especially lung cancer. PAHs can adhere to fine carbon particles in the atmosphere and thus penetrate our bodies primarily through breathing. Long-term exposure to PAH-containing air in polluted cities was found to increase the risk of lung cancer deaths. Aside from PAHs and other fine carbon particles, another environmental pollutant, nitric oxide, was found to increase the risk of lung cancer in a European population of nonsmokers. Other studies have shown that nitric oxide can induce lung cancer and promote metastasis. The increased risk of childhood leukemia associated with exposure to motor vehicle exhaust was also reported (64).
Indoor air pollutants such as volatile organic compounds and pesticides increase the risk of childhood leukemia and lymphoma, and children as well as adults exposed to pesticides have increased risk of brain tumors, Wilm’s tumors, Ewing’s sarcoma, and germ cell tumors. In utero exposure to environmental organic pollutants was found to increase the risk for testicular cancer. In addition, dioxan, an environmental pollutant from incinerators, was found to increase the risk of sarcoma and lymphoma.
Long-term exposure to chlorinated drinking water has been associated with increased risk of cancer. Nitrates, in drinking water, can transform to mutagenic N-nitroso compounds, which increase the risk of lymphoma, leukemia, colorectal cancer, and bladder cancer (64).
Radiation
Up to 10% of total cancer cases may be induced by radiation (64), both ionizing and non-ionizing, typically from radioactive substances and ultraviolet (UV), pulsed electromagnetic fields. Cancers induced by radiation include some types of leukemia, lymphoma, thyroid cancers, skin cancers, sarcomas, lung and breast carcinomas. One of the best examples of increased risk of cancer after exposure to radiation is the increased incidence of total malignancies observed in Sweden after exposure to radioactive fallout from the Chernobyl nuclear power plant. Radon and radon decay products in the home and/or at workplaces (such as mines) are the most common sources of exposure to ionizing radiation. The presence of radioactive nuclei from radon, radium, and uranium was found to increase the risk of gastric cancer in rats. Another source of radiation exposure is x-rays used in medical settings for diagnostic or therapeutic purposes. In fact, the risk of breast cancer from x-rays is highest among girls exposed to chest irradiation at puberty, a time of intense breast development. Other factors associated with radiation-induced cancers in humans are patient age and physiological state, synergistic interactions between radiation and carcinogens, and genetic susceptibility toward radiation.

Non-ionizing radiation derived primarily from sunlight includes UV rays, which are carcinogenic to humans. Exposure to UV radiation is a major risk for various types of skin cancers including basal cell carcinoma, squamous cell carcinoma, and melanoma. Along with UV exposure from sunlight, UV exposure from sun beds for cosmetic tanning may account for the growing incidence of melanoma. Depletion of the ozone layer in the stratosphere can augment the dose-intensity of UVB and UVC, which can further increase the incidence of skin cancer.
Low-frequency electromagnetic fields can cause clastogenic DNA damage. The sources of electromagnetic field exposure are high-voltage power lines, transformers, electric train engines, and more generally, all types of electrical equipments. An increased risk of cancers such as childhood leukemia, brain tumors and breast cancer has been attributed to electromagnetic field exposure. For instance, children living within 200 m of high-voltage power lines have a relative risk of leukemia of 69%, whereas those living between 200 and 600 m from these power lines have a relative risk of 23%. In addition, a recent meta-analysis of all available epidemiologic data showed that daily prolonged use of mobile phones for 10 years or more showed a consistent pattern of an increased risk of brain tumors (64).
Fruit, Vegetables, Spices, Condiments and Cereals with the Potential to Prevent & Reverse Cancer

Fruit, Vegetables, Spices, Condiments and Cereals with the Potential Increase Interstitial Fluid Acidosis and Cause Cancer

www.phmiracleproducts.com

Lecture in Dubai – The Annual Conference on Bacterial, Viral and Infectious Diseases
Join Robert O Young PhD and Galina Migalko MD in Dubai on December 5th and 6th, 2018 for the Annual Conference on Bacterial, Viral and Infectious Diseases. They will be Key Note Speakers and doing a workshop on the New Biology.

For more information and to register go to: https://bacterialdiseases.infectiousconferences.com/organizing-committee.php
The following is the abstract for Dr. Young’s lecture:
The Dismantling of the Viral Theory
Robert O Young CPT, MSc, DSc, PhD, Naturopathic Practitioner

Abstract
There is now over 100 years of documented history and research on the Polio virus and whether or not its treatment by inoculation has been successful in eradicating Polio. I am suggesting in this article and in my lecture that there are significant findings based on historical and past and current research, including my own that the viral theory of Polio and possibly other modern-day diseases, such as Post-Polio Syndrome, Polio Vaccine-Induced Paralysis, Legionnaires, CNS disease, Cancer, HIV/AIDS and now Zika may be caused by acidic chemical poisoning from DDT (dichloro-diphenyl-trichloroethane) and other related DDT pesticides, acidic vaccinations, and other factors including lifestyle and dietary factors rather than from a lone infectious virus. I will present ten historical graphs outlining the history of Polio, the production of DDT, BHC, Lead, Arsenic, Polio vaccinations and the author’s theory that chemical poisoning, vaccination, and lifestyle and dietary choices are a more likely causes for the symptoms of Polio, neurological diseases, Cancer, HIV/AIDS , Zika and NOW the Coronavirus.
https://www.linkedin.com/pulse/lecture-dubai-annual-conference-bacterial-viral-infectious-young/https://bacterialdiseases.infectiousconferences.com/organizing-committee.php

THE POSSIBLE CAUSE OF POLIO, POST-POLIO, CNS, PVIPD, LEGIONNAIRES, AIDS and the CANCER EPIDEMIC – MASS ACIDIC CHEMICAL POISONING?

References
1. L. N. Kolonel, D. Altshuler, and B. E. Henderson. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat. Rev. Cancer. 4:519–27 (2004) doi:10.1038/nrc1389. [PubMed]
2. J. K. Wiencke. Impact of race/ethnicity on molecular pathways in human cancer. Nat. Rev. Cancer. 4:79–84 (2004) doi:10.1038/nrc1257. [PubMed]
3. R. G. Ziegler, R. N. Hoover, M. C. Pike, A. Hildesheim, A. M. Nomura, D. W. West, A. H. Wu-Williams, L. N. Kolonel, P. L. Horn-Ross, J. F. Rosenthal, and M. B. Hyer. Migration patterns and breast cancer risk in Asian-American women. J. Natl. Cancer Inst.85:1819–27 (1993) doi:10.1093/jnci/85.22.1819. [PubMed]
4. W. Haenszel and M. Kurihara. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J. Natl. Cancer Inst.40:43–68 (1968). [PubMed]
5. A. S. Hamilton and T. M. Mack. Puberty and genetic susceptibility to breast cancer in a case-control study in twins. N. Engl. J. Med.348:2313–22 (2003) doi:10.1056/NEJMoa021293. [PubMed]
6. A. Jemal, R. Siegel, E. Ward, T. Murray, J. Xu, and M. J. Thun. Cancer statistics, 2007. CA Cancer J. Clin.57:43–66 (2007). [PubMed]
7. F. Brayand, and B. Moller. Predicting the future burden of cancer. Nat. Rev. Cancer. 6:63–74 (2006) doi:10.1038/nrc1781. [PubMed]
8. P. Lichtenstein, N. V. Holm, P. K. Verkasalo, A. Iliadou, J. Kaprio, M. Koskenvuo, E. Pukkala, A. Skytthe, and K. Hemminki. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med.343:78–85 (2000) doi:10.1056/NEJM200007133430201. [PubMed]
9. K. R. Loeb, and L. A. Loeb. Significance of multiple mutations in cancer. Carcinogenesis. 21:379–85 (2000) doi:10.1093/carcin/21.3.379. [PubMed]
10. W. C. Hahn, and R. A. Weinberg. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer. 2:331–41 (2002) doi: 10.1038/nrc795. [PubMed]
11. L. A. Mucci, S. Wedren, R. M. Tamimi, D. Trichopoulos, and H. O. Adami. The role of gene-environment interaction in the aetiology of human cancer: examples from cancers of the large bowel, lung and breast. J. Intern. Med.249:477–93 (2001) doi:10.1046/j.1365-2796.2001.00839.x. [PubMed]
12. K. Czene, and K. Hemminki. Kidney cancer in the Swedish Family Cancer Database: familial risks and second primary malignancies. Kidney Int.61:1806–13 (2002) doi:10.1046/j.1523-1755.2002.00304.x.[PubMed]
13. P. Irigaray, J. A. Newby, R. Clapp, L. Hardell, V. Howard, L. Montagnier, S. Epstein, and D. Belpomme. Lifestyle-related factors and environmental agents causing cancer: an overview. Biomed. Pharmacother.61:640–58 (2007) doi:10.1016/j.biopha.2007.10.006. [PubMed]
14. M. F. Denissenko, A. Pao, M. Tang, and G. P. Pfeifer. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 274:430–2 (1996) doi:10.1126/science.274.5286.430.[PubMed]
15. R. J. Anto, A. Mukhopadhyay, S. Shishodia, C. G. Gairola, and B. B. Aggarwal. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): correlation with induction of cyclooxygenase-2. Carcinogenesis. 23:1511–8 (2002) doi: 10.1093/carcin/23.9.1511. [PubMed]
16. S. Shishodiaand, and B. B. Aggarwal. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-kappaB by suppressing activation of IkappaBalpha kinase in human non-small cell lung carcinoma: correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res. 64:5004–12 (2004) doi:10.1158/0008-5472.CAN-04-0206. [PubMed]
17. H. Ichikawa, Y. Nakamura, Y. Kashiwada, and B. B. Aggarwal. Anticancer drugs designed by mother nature: ancient drugs but modern targets. Curr Pharm Des. 13:3400–16 (2007) doi:10.2174/138161207782360500. [PubMed]
18. A. J. Tuyns. Epidemiology of alcohol and cancer. Cancer Res. 39:2840–3 (1979). [PubMed]
19. H. Maier, E. Sennewald, G. F. Heller, and H. Weidauer. Chronic alcohol consumption—the key risk factor for pharyngeal cancer. Otolaryngol. Head Neck Surg.110:168–73 (1994). [PubMed]
20. H. K. Seitz, F. Stickel, and N. Homann. Pathogenetic mechanisms of upper aerodigestive tract cancer in alcoholics. Int. J. Cancer. 108:483–7 (2004) doi:10.1002/ijc.11600. [PubMed]
21. R. Doll, and R. Peto. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 66:1191–308 (1981). [PubMed]
22. R. R. Williams, and J. W. Horm. Association of cancer sites with tobacco and alcohol consumption and socioeconomic status of patients: interview study from the Third National Cancer Survey. J. Natl. Cancer Inst.58:525–47 (1977). [PubMed]
23. N. Hamajima et al. Alcohol, tobacco and breast cancer—collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br. J. Cancer. 87:1234–45 (2002) doi:10.1038/sj.bjc.6600596. [PMC free article] [PubMed]
24. M. P. Longnecker, P. A. Newcomb, R. Mittendorf, E. R. Greenberg, R. W. Clapp, G. F. Bogdan, J. Baron, B. MacMahon, and W. C. Willett. Risk of breast cancer in relation to lifetime alcohol consumption. J. Natl. Cancer Inst.87:923–9 (1995) doi:10.1093/jnci/87.12.923. [PubMed]
25. F. Stickel, D. Schuppan, E. G. Hahn, and H. K. Seitz. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut. 51:132–9 (2002) doi:10.1136/gut.51.1.132. [PMC free article] [PubMed]
26. H. K. Seitz, G. Poschl, and U. A. Simanowski. Alcohol and cancer. Recent Dev Alcohol. 14:67–95 (1998) doi:10.1007/0-306-47148-5_4. [PubMed]
27. H. K. Seitz, S. Matsuzaki, A. Yokoyama, N. Homann, S. Vakevainen, and X. D. Wang. Alcohol and cancer. Alcohol Clin. Exp. Res.25:137S–143S (2001). [PubMed]
28. F. Donato, U. Gelatti, R. M. Limina, and G. Fattovich. Southern Europe as an example of interaction between various environmental factors: a systematic review of the epidemiologic evidence. Oncogene. 25:3756–70 (2006) doi:10.1038/sj.onc.1209557. [PubMed]
29. G. Poschl, and H. K. Seitz. Alcohol and cancer. Alcohol Alcohol. 39:155–65 (2004) doi:10.1093/alcalc/agh057. [PubMed]
30. G. Szabo, P. Mandrekar, S. Oak, and J. Mayerle. Effect of ethanol on inflammatory responses. Implications for pancreatitis. Pancreatology. 7:115–23 (2007) doi:10.1159/000104236. [PMC free article][PubMed]
31. B. B. Aggarwal. Nuclear factor-kappaB: the enemy within. Cancer Cell. 6:203–208 (2004) doi:10.1016/j.ccr.2004.09.003. [PubMed]
32. M. Kuratsune, S. Kohchi, and A. Horie. Carcinogenesis in the esophagus. I. Penetration of benzo(a) pyrene and other hydrocarbons into the esophageal mucosa. Gann. 56:177–87 (1965). [PubMed]
33. C. La Vecchia, A. Tavani, S. Franceschi, F. Levi, G. Corrao, and E. Negri. Epidemiology and prevention of oral cancer. Oral Oncol.33:302–312 (1997). [PubMed]
34. P. Boffetta, M. Hashibe, C. La Vecchia, W. Zatonski, and J. Rehm. The burden of cancer attributable to alcohol drinking. Int. J. Cancer. 119:884–887 (2006) doi:10.1002/ijc.21903. [PubMed]
35. W. C. Willett. Diet and cancer. Oncologist. 5:393–404 (2000) doi:10.1634/theoncologist.5-5-393.[PubMed]
36. S. A. Bingham, R. Hughes, and A. J. Cross. Effect of white versus red meat on endogenous N-nitrosation in the human colon and further evidence of a dose response. J. Nutr.132:3522S–3525S (2002).[PubMed]
37. A. Chao, M. J. Thun, C. J. Connell, M. L. McCullough, E. J. Jacobs, W. D. Flanders, C. Rodriguez, R. Sinha, and E. E. Calle. Meat consumption and risk of colorectal cancer. JAMA. 293:172–182 (2005) doi:10.1001/jama.293.2.172. [PubMed]
38. N. Hogg. Red meat and colon cancer: heme proteins and nitrite in the gut. A commentary on diet-induced endogenous formation of nitroso compounds in the GI tract. Free Radic. Biol. Med.43:1037–1039 (2007) doi:10.1016/j.freeradbiomed.2007.07.006. [PubMed]
39. C. Rodriguez, M. L. McCullough, A. M. Mondul, E. J. Jacobs, A. Chao, A. V. Patel, M. J. Thun, and E. E. Calle. Meat consumption among Black and White men and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol. Biomarkers Prev. 15:211–216 (2006) doi:10.1158/1055-9965.EPI-05-0614. [PubMed]
40. R. Garcia-Closas, M. Garcia-Closas, M. Kogevinas, N. Malats, D. Silverman, C. Serra, A. Tardon, A. Carrato, G. Castano-Vinyals, M. Dosemeci, L. Moore, N. Rothman, and R. Sinha. Food, nutrient and heterocyclic amine intake and the risk of bladder cancer. Eur. J. Cancer. 43:1731–1740 (2007) doi:10.1016/j.ejca.2007.05.007. [PubMed]
41. A. Tappel. Heme of consumed red meat can act as a catalyst of oxidative damage and could initiate colon, breast and prostate cancers, heart disease and other diseases. Med. Hypotheses. 68:562–4 (2007) doi:10.1016/j.mehy.2006.08.025. [PubMed]
42. L. H. O’Hanlon. High meat consumption linked to gastric-cancer risk. Lancet Oncol. 7:287 (2006) doi:10.1016/S1470-2045(06)70638-6. [PubMed]
43. T. N. Toporcov, J. L. Antunes, and M. R. Tavares. Fat food habitual intake and risk of oral cancer. Oral Oncol. 40:925–931 (2004) doi:10.1016/j.oraloncology.2004.04.007. [PubMed]
44. O. Dosil-Diaz, A. Ruano-Ravina, J. J. Gestal-Otero, and J. M. Barros-Dios. Meat and fish consumption and risk of lung cancer: A case-control study in Galicia, Spain. Cancer Lett.252:115–122 (2007) doi:10.1016/j.canlet.2006.12.008. [PubMed]
45. S. N. Lauber, and N. J. Gooderham. The cooked meat derived genotoxic carcinogen 2-amino-3-methylimidazo[4,5-b]pyridine has potent hormone-like activity: mechanistic support for a role in breast cancer. Cancer Res.67:9597–0602 (2007) doi:10.1158/0008–5472.CAN-07-1661. [PubMed]
46. D. Divisi, S. Di Tommaso, S. Salvemini, M. Garramone, and R. Crisci. Diet and cancer. Acta Biomed. 77:118–123 (2006). [PubMed]
47. Y. F. Sasaki, S. Kawaguchi, A. Kamaya, M. Ohshita, K. Kabasawa, K. Iwama, K. Taniguchi, and S. Tsuda. The comet assay with 8 mouse organs: results with 39 currently used food additives. Mutat. Res.519:103–119 (2002). [PubMed]
48. M. Durando, L. Kass, J. Piva, C. Sonnenschein, A. M. Soto, E. H. Luque, and M. Munoz-de-Toro. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ. Health Perspect.115:80–6 (2007). [PMC free article] [PubMed]
49. S. M. Ho, W. Y. Tang, J. Belmonte de Frausto, and G. S. Prins. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res.66:5624–32 (2006) doi:10.1158/0008-5472.CAN-06-0516.[PMC free article] [PubMed]
50. A. Szymanska-Chabowska, J. Antonowicz-Juchniewicz, and R. Andrzejak. Some aspects of arsenic toxicity and carcinogenicity in living organism with special regard to its influence on cardiovascular system, blood and bone marrow. Int. J. Occup. Med. Environ. Health. 15:101–116 (2002). [PubMed]
51. E. E. Calle, C. Rodriguez, K. Walker-Thurmond, and M. J. Thun. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 348:1625–1638 (2003) doi:10.1056/NEJMoa021423. [PubMed]
52. A. Drewnowski, and B. M. Popkin. The nutrition transition: new trends in the global diet. Nutr. Rev.55:31–43 (1997). [PubMed]
53. S. D. Hursting, L. M. Lashinger, L. H. Colbert, C. J. Rogers, K. W. Wheatley, N. P. Nunez, S. Mahabir, J. C. Barrett, M. R. Forman, and S. N. Perkins. Energy balance and carcinogenesis: underlying pathways and targets for intervention. Curr. Cancer Drug Targets. 7:484–491 (2007) doi:10.2174/156800907781386623. [PubMed]
54. A. Nareika, Y. B. Im, B. A. Game, E. H. Slate, J. J. Sanders, S. D. London, M. F. Lopes-Virella, and Y. Huang. High glucose enhances lipopolysaccharide-stimulated CD14 expression in U937 mononuclear cells by increasing nuclear factor kappaB and AP-1 activities. J. Endocrinol.196:45–55 (2008) doi:10.1677/JOE-07-0145. [PubMed]
55. C. H. Tang, Y. C. Chiu, T. W. Tan, R. S. Yang, and W. M. Fu. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J. Immunol.179:5483–5492 (2007). [PubMed]
56. P. Pisani, D. M. Parkin, N. Munoz, and J. Ferlay. Cancer and infection: estimates of the attributable fraction in 1990. Cancer Epidemiol. Biomarkers Prev.6:387–400 (1997). [PubMed]
57. D. M. Parkin. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer. 118:3030–3044 (2006) doi:10.1002/ijc.21731. [PubMed]
58. S. Song, H. C. Pitot, and P. F. Lambert. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J. Virol.73:5887–5893 (1999). [PMC free article] [PubMed]
59. B. S. Blumberg, B. Larouze, W. T. London, B. Werner, J. E. Hesser, I. Millman, G. Saimot, and M. Payet. The relation of infection with the hepatitis B agent to primary hepatic carcinoma. Am. J. Pathol.81:669–682 (1975). [PMC free article] [PubMed]
60. T. M. Hagen, S. Huang, J. Curnutte, P. Fowler, V. Martinez, C. M. Wehr, B. N. Ames, and F. V. Chisari. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc. Natl. Acad. Sci. U S A. 91:12808–12812 (1994) doi:10.1073/pnas.91.26.12808. [PMC free article] [PubMed]
61. A. L. Jackson, and L. A. Loeb. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res.477:7–21 (2001) doi:10.1016/S0027-5107(01)00091-4. [PubMed]
62. N. De Maria, A. Colantoni, S. Fagiuoli, G. J. Liu, B. K. Rogers, F. Farinati, D. H. Van Thiel, and R. A. Floyd. Association between reactive oxygen species and disease activity in chronic hepatitis C. Free Radic. Biol. Med.21:291–5 (1996) doi:10.1016/0891–5849(96)00044-5. [PubMed]
63. K. Koike, T. Tsutsumi, H. Fujie, Y. Shintani, and M. Kyoji. Molecular mechanism of viral hepatocarcinogenesis. Oncology. 62(Suppl 1):29–37 (2002) doi:10.1159/000048273. [PubMed]
64. D. Belpomme, P. Irigaray, L. Hardell, R. Clapp, L. Montagnier, S. Epstein, and A. J. Sasco. The multitude and diversity of environmental carcinogens. Environ. Res.105:414–429 (2007) doi:10.1016/j.envres.2007.07.002. [PubMed]
65. Y. S. Guan, Q. He, M. Q. Wang, and P. Li. Nuclear factor kappa B and hepatitis viruses. Expert Opin. Ther. Targets. 12:265–280 (2008) doi:10.1517/14728222.12.3.265. [PubMed]
66. S. Takayama, H. Takahashi, Y. Matsuo, Y. Okada, and T. Manabe. Effects of Helicobacter pylori infection on human pancreatic cancer cell line. Hepatogastroenterology. 54:2387–2391 (2007). [PubMed]
67. K. A. Steinmetz, and J. D. Potter. Vegetables, fruit, and cancer prevention: a review. J. Am. Diet Assoc.96:1027–1039 (1996) doi:10.1016/S0002–8223(96)00273-8. [PubMed]
68. P. Greenwald. Lifestyle and medical approaches to cancer prevention. Recent Results Cancer Res.166:1–15 (2005). [PubMed]
69. H. Vainio, and E. Weiderpass. Fruit and vegetables in cancer prevention. Nutr. Cancer. 54:111–42 (2006) doi:10.1207/s15327914nc5401_13. [PubMed]
70. L. W. Wattenberg. Chemoprophylaxis of carcinogenesis: a review. Cancer Res. 26:1520–1526 (1966).[PubMed]
71. B. B. Aggarwal, and S. Shishodia. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol.71:1397–1421 (2006) doi:10.1016/j.bcp.2006.02.009. [PubMed]
72. H. Nishino, M. Murakosh, T. Ii, M. Takemura, M. Kuchide, M. Kanazawa, X. Y. Mou, S. Wada, M. Masuda, Y. Ohsaka, S. Yogosawa, Y. Satomi, and K. Jinno. Carotenoids in cancer chemoprevention. Cancer Metastasis Rev.21:257–264 (2002) doi:10.1023/A:1021206826750. [PubMed]
73. K. B. Harikumar, and B. B. Aggarwal. Resveratrol: A multitargeted agent for age-associated chronic diseases. Cell Cycle. 7:1020–1037 (2008). [PubMed]
74. G. L. Russo. Ins and outs of dietary phytochemicals in cancer chemoprevention. Biochem. Pharmacol. 74:533–544 (2007) doi:10.1016/j.bcp.2007.02.014. [PubMed]
75. R. Agarwal, C. Agarwal, H. Ichikawa, R. P. Singh, and B. B. Aggarwal. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 26:4457–98 (2006). [PubMed]
76. E. G. Rogan. The natural chemopreventive compound indole-3-carbinol: state of the science. In Vivo. 20:221–228 (2006). [PubMed]
77. N. Juge, R. F. Mithen, and M. Traka. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 64:1105–27 (2007) doi:10.1007/s00018-007-6484-5. [PubMed]
78. L. Chen, and H. Y. Zhang. Cancer preventive mechanisms of the green tea polyphenol (−)-epigallocatechin-3-gallate. Molecules. 12:946–957 (2007). [PMC free article] [PubMed]
79. P. Anand, C. Sundaram, S. Jhurani, A. B. Kunnumakkara, and B. B. Aggarwal. Curcumin and cancer: An “old-age” disease with an “age-old” solution. Cancer Lett. in press (2008). [PubMed]
80. F. Khanum, K. R. Anilakumar, and K. R. Viswanathan. Anticarcinogenic properties of garlic: a review. Crit. Rev. Food Sci. Nutr.44:479–488 (2004) doi:10.1080/10408690490886700. [PubMed]
81. G. Sethi, K. S. Ahn and B. B. Aggarwal. Targeting NF-kB activation pathway by thymoquinone: Role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mole Cancer Res. in press (2008). [PubMed]
82. Y. J. Surh. Anti-tumor promoting potential of selected spice ingredients with antioxidative and anti-inflammatory activities: a short review. Food Chem. Toxicol.40:1091–1097 (2002) doi:10.1016/S0278-6915(02)00037-6. [PubMed]
83. Y. Shukla, and M. Singh. Cancer preventive properties of ginger: a brief review. Food Chem. Toxicol.45:683–690 (2007) doi:10.1016/j.fct.2006.11.002. [PubMed]
84. M. M. al-Harbi, S. Qureshi, M. Raza, M. M. Ahmed, A. B. Giangreco, and A. H. Shah. Influence of anethole treatment on the tumour induced by Ehrlich ascites carcinoma cells in paw of Swiss albino mice. Eur. J. Cancer Prev.4:307–318 (1995) doi:10.1097/00008469-199508000-00006. [PubMed]
85. C. K. Sen, K. E. Traber, and L. Packer. Inhibition of NF-kappa B activation in human T-cell lines by anetholdithiolthione. Biochem. Biophys. Res. Commun.218:148–53 (1996) doi:10.1006/bbrc.1996.0026.[PubMed]
86. R. A. Lubet, V. E. Steele, I. Eto, M. M. Juliana, G. J. Kelloff, and C. J. Grubbs. Chemopreventive efficacy of anethole trithione, N-acetyl-L-cysteine, miconazole and phenethylisothiocyanate in the DMBA-induced rat mammary cancer model. Int. J. Cancer. 72:95–101 (1997) doi:10.1002/(SICI)1097-0215(19970703)72:1<95::AID-IJC14>3.0.CO;2-9. [PubMed]
87. Y. Nakagawa, and T. Suzuki. Cytotoxic and xenoestrogenic effects via biotransformation of trans-anethole on isolated rat hepatocytes and cultured MCF-7 human breast cancer cells. Biochem. Pharmacol.66:63–73 (2003) doi:10.1016/S0006-2952(03)00208-9. [PubMed]
88. S. Lam, C. MacAulay, J. C. Le Riche, Y. Dyachkova, A. Coldman, M. Guillaud, E. Hawk, M. O. Christen, and A. F. Gazdar. A randomized phase IIb trial of anethole dithiolethione in smokers with bronchial dysplasia. J. Natl. Cancer Inst.94:1001–1009 (2002). [PubMed]
89. S. Shishodia, and B. B. Aggarwal. Diosgenin inhibits osteoclastogenesis, invasion, and proliferation through the downregulation of Akt, I kappa B kinase activation and NF-kappa B-regulated gene expression. Oncogene. 25:1463–1473 (2006) doi:10.1038/sj.onc.1209194. [PubMed]
90. R. Ghosh, N. Nadiminty, J. E. Fitzpatrick, W. L. Alworth, T. J. Slaga, and A. P. Kumar. Eugenol causes melanoma growth suppression through inhibition of E2F1 transcriptional activity. J. Biol. Chem.280:5812–5819 (2005) doi:10.1074/jbc.M411429200. [PubMed]
91. K. Sukumaran, M. C. Unnikrishnan, and R. Kuttan. Inhibition of tumour promotion in mice by eugenol. Indian J. Physiol. Pharmacol.38:306–308 (1994). [PubMed]
92. K. Imaida, M. Hirose, S. Yamaguchi, S. Takahashi, and N. Ito. Effects of naturally occurring antioxidants on combined 1,2-dimethylhydrazine- and 1-methyl-1-nitrosourea-initiated carcinogenesis in F344 male rats. Cancer Lett.55:53–59 (1990) doi:10.1016/0304-3835(90)90065-6. [PubMed]
93. M. Pisano, G. Pagnan, M. Loi, M. E. Mura, M. G. Tilocca, G. Palmieri, D. Fabbri, M. A. Dettori, G. Delogu, M. Ponzoni, and C. Rozzo. Antiproliferative and pro-apoptotic activity of eugenol-related biphenyls on malignant melanoma cells. Mol Cancer. 6:8 (2007) doi:10.1186/1476-4598-6-8.[PMC free article] [PubMed]
94. S. S. Kim, O. J. Oh, H. Y. Min, E. J. Park, Y. Kim, H. J. Park, Y. Nam Han, and S. K. Lee. Eugenol suppresses cyclooxygenase-2 expression in lipopolysaccharide-stimulated mouse macrophage RAW264.7 cells. Life Sci. 73:337–348 (2003) doi:10.1016/S0024–3205(03)00288-1. [PubMed]
95. H. P. Deigner, G. Wolf, U. Ohlenmacher, and J. Reichling. 1¢-Hydroxyeugenol- and coniferyl alcohol derivatives as effective inhibitors of 5-lipoxygenase and Cu(2+)-mediated low density lipoprotein oxidation. Evidence for a dual mechanism. Arzneimittelforschung. 44:956–961 (1994). [PubMed]
96. C. J. Rompelberg, M. J. Steenwinkel, J. G. van Asten, J. H. van Delft, R. A. Baan, and H. Verhagen. Effect of eugenol on the mutagenicity of benzo[a]pyrene and the formation of benzo[a]pyrene-DNA adducts in the lambda-lacZ-transgenic mouse. Mutat. Res.369:87–96 (1996) doi:10.1016/S0165-1218(96)90052-X. [PubMed]
97. D. P. Richardson. The grain, the wholegrain and nothing but the grain: the science behind wholegrain and the reduced risk of heart disease and cancer. Nutr. Bull.25:353–360 (2000) doi:10.1046/j.1467-3010.2000.00083.x.
98. H. E. Miller, F. Rigelhof, L. Marquart, A. Prakash, and M. Kanter. Antioxidant content of whole grain breakfast cereals, fruits and vegetables. J. Am. Coll. Nutr.19:312S–319S (2000). [PubMed]
99. J. L. Slavin, D. Jacobs, and L. Marquart. Grain processing and nutrition. Crit. Rev. Food Sci. Nutr.40:309–326 (2000) doi:10.1080/10408690091189176. [PubMed]
100. L. Chatenoud, A. Tavani, C. La Vecchia, D. R. Jacobs, Jr, E. Negri, F. Levi, and S. Franceschi. Whole grain food intake and cancer risk. Int. J. Cancer. 77:24–8 (1998) doi:10.1002/(SICI)1097-0215(19980703)77:1<24::AID-IJC5>3.0.CO;2-1. [PubMed]
101. D. R. Jacobs, Jr, L. Marquart, J. Slavin, and L. H. Kushi. Whole-grain intake and cancer: an expanded review and meta-analysis. Nutr. Cancer. 30:85–96 (1998). [PubMed]
102. L. Marquart, K. L. Wiemer, J. M. Jones, and B. Jacob. Whole grains health claims in the USA and other efforts to increase whole-grain consumption. Proc. Nutr. Soc.62:151–160 (2003) doi:10.1079/PNS2003242. [PubMed]
103. M. Eastwood, and D. Kritchevsky. Dietary fiber: how did we get where we are? Annu. Rev. Nutr.25:1–8 (2005) doi:10.1146/annurev.nutr.25.121304.131658. [PubMed]
104. A. McIntyre, P. R. Gibson, and G. P. Young. Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut. 34:386–391 (1993) doi:10.1136/gut.34.3.386.[PMC free article] [PubMed]
105. J. L. Slavin, D. Jacobs, L. Marquart, and K. Wiemer. The role of whole grains in disease prevention. J. Am. Diet Assoc.101:780–5 (2001) doi:10.1016/S0002-8223(01)00194-8. [PubMed]
106. K. S. Ahn, G. Sethi, K. Krishnan, and B. B. Aggarwal. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J. Biol. Chem.282:809–820 (2007) doi:10.1074/jbc.M610028200. [PubMed]
107. F. H. Sarkar, S. Adsule, S. Padhye, S. Kulkarni, and Y. Li. The role of genistein and synthetic derivatives of isoflavone in cancer prevention and therapy. Mini Rev. Med. Chem.6:401–407 (2006) doi:10.2174/138955706776361439. [PubMed]
108. K. W. Lee, H. J. Lee, Y. J. Surh, and C. Y. Lee. Vitamin C and cancer chemoprevention: reappraisal. Am. J. Clin. Nutr.78:1074–1078 (2003). [PubMed]
109. B. A. Ingraham, B. Bragdon, and A. Nohe. Molecular basis of the potential of vitamin D to prevent cancer. Curr. Med. Res. Opin.24:139–149 (2008) doi:10.1185/030079907X253519. [PubMed]
110. F. W. Booth, M. V. Chakravarthy, S. E. Gordon, and E. E. Spangenburg. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J. Appl. Physiol.93:3–30 (2002).[PubMed]
111. G. A. Colditz, C. C. Cannuscio, and A. L. Frazier. Physical activity and reduced risk of colon cancer: implications for prevention. Cancer Causes Control. 8:649–67 (1997) doi:10.1023/A:1018458700185.[PubMed]
112. A. R. Shors, C. Solomon, A. McTiernan, and E. White. Melanoma risk in relation to height, weight, and exercise (United States). Cancer Causes Control. 12:599–606 (2001) doi:10.1023/A:1011211615524.[PubMed]
113. A. Tannenbaum, and H. Silverstone. The initiation and growth of tumors. Introduction. I. Effects of underfeeding. Am. J. Cancer. 38:335–350 (1940).
114. S. D. Hursting, J. A. Lavigne, D. Berrigan, S. N. Perkins, and J. C. Barrett. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu. Rev. Med.54:131–152 (2003) doi:10.1146/annurev.med.54.101601.152156. [PubMed]
115. M. H. Ross, and G. Bras. Lasting influence of early caloric restriction on prevalence of neoplasms in the rat. J. Natl. Cancer Inst.47:1095–1113 (1971). [PubMed]
116. D. Albanes. Total calories, body weight, and tumor incidence in mice. Cancer Res.47:1987–92 (1987).[PubMed]
117. L. Gross, and Y. Dreyfuss. Reduction in the incidence of radiation-induced tumors in rats after restriction of food intake. Proc. Natl. Acad. Sci. U S A. 81:7596–7598 (1984) doi:10.1073/pnas.81.23.7596. [PMC free article] [PubMed]
118. L. Gross, and Y. Dreyfuss. Prevention of spontaneous and radiation-induced tumors in rats by reduction of food intake. Proc. Natl. Acad. Sci. U S A. 87:6795–6797 (1990) doi:10.1073/pnas.87.17.6795.[PMC free article] [PubMed]
119. K. Yoshida, T. Inoue, K. Nojima, Y. Hirabayashi, and T. Sado. Calorie restriction reduces the incidence of myeloid leukemia induced by a single whole-body radiation in C3H/He mice. Proc. Natl. Acad. Sci. U S A. 94:2615–2619 (1997) doi:10.1073/pnas.94.6.2615. [PMC free article] [PubMed]
120. V. D. Longo, and C. E. Finch. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science. 299:1342–1346 (2003) doi:10.1126/science.1077991. [PubMed]


